Section 4 Cross-disease comparisons
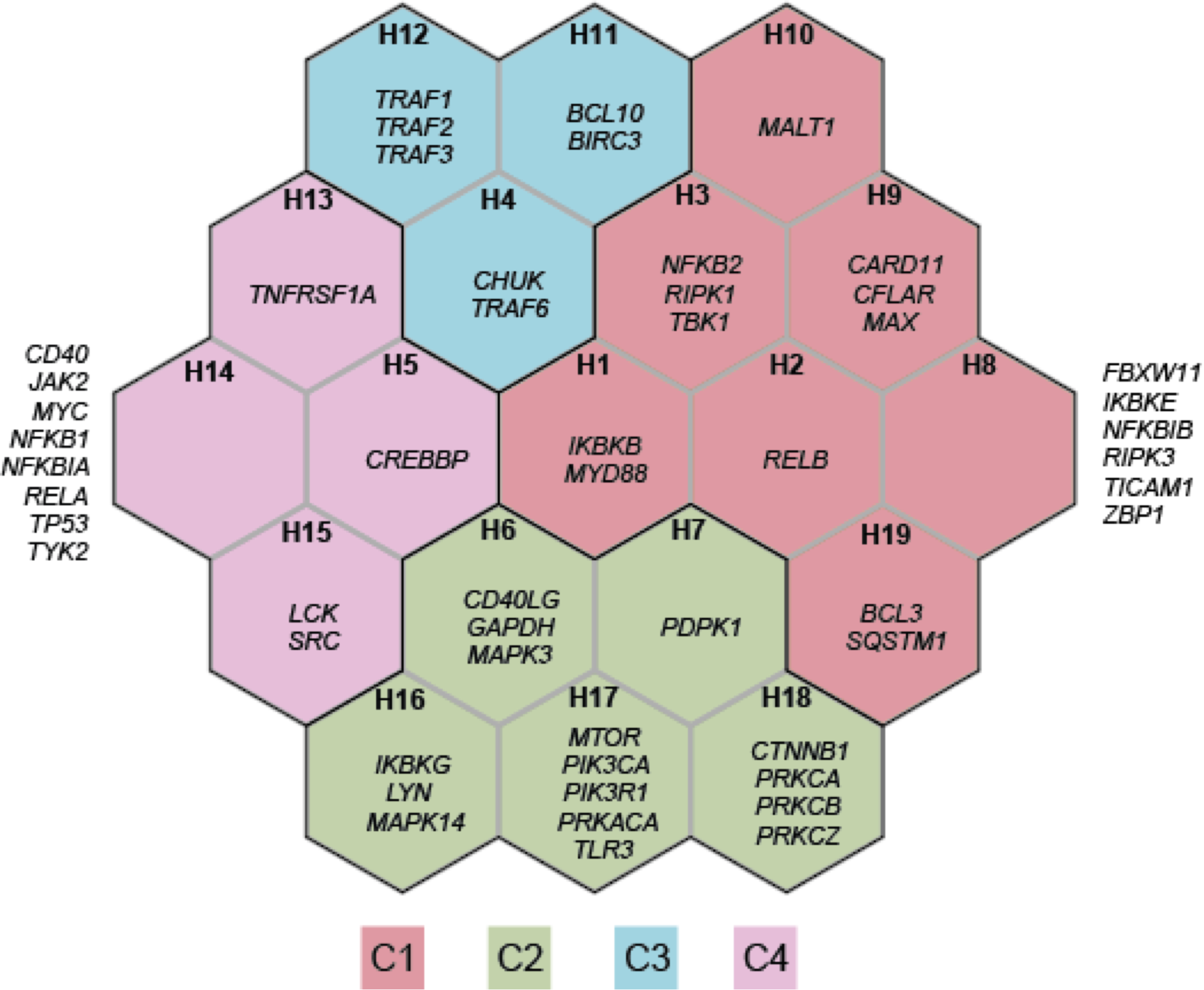
Figure 4.1: Comparisons using the supra-hexagonal map. This map was learned from the prioritisation information of 53 crosstalk genes in kidney stone disease (KSD) and 8 immune-related traits. Each map illustrates a trait-specific crosstalk gene prioritisation profile. Across traits, genes with similar prioritisation patterns are mapped onto the same or nearly position in the map. The outermost frame represents the landscape for the traits analysed, from which geometric locations of traits delineate their relationships (by the similarity of prioritisation profiles between traits). AS: Ankylosing Spondylitis; CRO: Crohn’s Disease; MS: Multiple Sclerosis; PSO: Psoriasis; RA: Rheumatoid Arthritis; SLE: Systemic Lupus Erythematosus; T1D: Type I Diabetes; UC: Ulcerative Colitis.
4.1 Do comparison
# read cross-disease prioritisation matrix
data.file <- file.path(RData.location, "crossdisease_matrix.txt")
data <- read_delim(data.file, delim='\t') %>% column_to_rownames('target')
# analysed using the self-organising learning algorithm
sMap <- data %>% sPipeline(scaling=1)
# the resulting map partitioned into gene clusters
sBase <- sMap %>% sDmatCluster()
# write into a file 'crossdisease_suprahex.txt'
sWriteData(sMap, data, sBase, keep.data=T) %>% transmute(Target=ID, Index=str_c('H',Hexagon_index), Cluster=str_c('C',Cluster_base), KSD,AS,PSO,UC,CRO,RA,MS,SLE,T1D) %>% write_delim('crossdisease_suprahex.txt', delim='\t')4.2 Clustered genes
Clustered genes identified above (crossdisease_suprahex.txt) can be explored: